About the scMagnify#
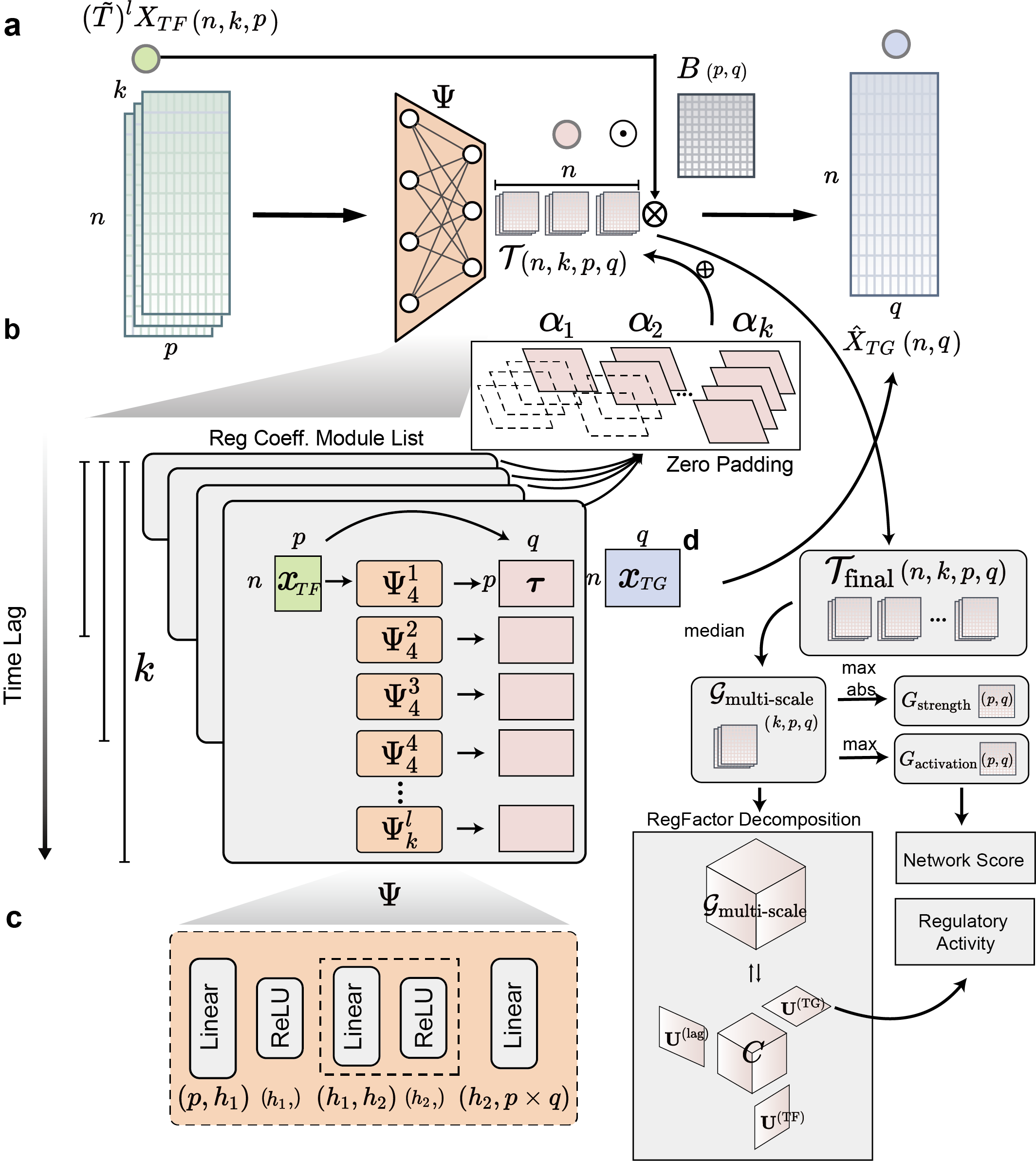
scMagnify leverages a deep-learning framework to reconstruct and decompose multi-scale dynamic gene regulatory networks (GRNs) from single-cell multi-omic (gene expression and chromatin accessibility) data. The inference is achieved through a nonlinear Granger causality model, implemented as an interpretable multi-scale neural network.
The scMagnify workflow begins by integrating three primary inputs:
A gene expression matrix (\(X\)).
A cell transition matrix (\(T\)), typically derived from pseudotime or trajectory inference, which defines a directed acyclic graph (DAG) of cellular relationships.
A basal TF binding network (\(B\)), which is constructed from peak-to-gene correlations and TF motif scanning. This network acts as a structural constraint, incorporating a priori biological knowledge from chromatin accessibility data.
To capture non-linear dynamics and temporal relationships, scMagnify employs a self-explaining neural network that functions as a non-linear extension of the classical Vector Autoregressive (VAR) framework. Instead of using simple time lags, the model uses a graph diffusion operator (\(\tilde{T}\)) derived from the cell transition matrix to aggregate TF expression information from \(k\)-hop ancestral cells in the trajectory graph.
The model’s core architecture (detailed in Supplementary Fig. 1) predicts the target gene (TG) expression \(\hat{X}_{TG}\) by integrating signals across multiple time scales (lags) using parallel branches, each weighted by a learnable attention mechanism \(\alpha_{k}\). The full model is formulated as:
Here, \(\Psi_{k}^{l}\) represents the neural network for a specific branch and lag, \((\tilde{T})^{l}X_{TF}\) is the graph-diffused TF expression from \(l\)-step ancestors, and \(\odot B\) is the element-wise application of the chromatin-derived structural constraint.
The model is trained by minimizing a penalized loss function using mini-batch gradient descent:
This loss combines the standard Mean Squared Error (\(\mathcal{L}_{MSE}\)) with two regularization terms: a Group Elastic Net penalty (\(\mathcal{L}_{sparsity}\)) to enforce GRN sparsity and a temporal smoothness penalty (\(\mathcal{L}_{smooth}\)) to ensure regulatory coefficients evolve smoothly along the trajectory.
After training, the model outputs a multi-scale regulatory coefficient tensor (\(\mathcal{J}_{total}\)). This tensor is filtered and aggregated (via median) to produce the final, robust multi-scale GRN, \(\mathcal{G}_{multi-scale} \in \mathbb{R}^{K\times P\times Q}\), which quantifies regulatory strengths across all time lags (\(K\)), TFs (\(P\)), and TGs (\(Q\)).
RegFactor Decomposition#
A key innovation of scMagnify is the systematic decomposition of the inferred multi-scale GRN to identify combinatorial regulatory logic. This is achieved by applying Tucker decomposition to the third-order \(\mathcal{G}_{multi-scale}\) tensor:
This decomposition deconstructs the network into a core tensor (\(C\)) and three factor matrices, which define:
Co-regulating TF modules (\(U^{(TF)}\))
Shared target gene (TG) modules (\(U^{(TG)}\))
Temporal activation profiles (\(U^{(lag)}\))
A specific combination of these three components, linked by the core tensor, constitutes a ‘RegFactor’. This allows for a hierarchical dissection of gene regulation, moving from individual regulators to dynamic, combinatorial modules.
Downstream Applications#
The scMagnify framework includes a suite of integrated downstream analyses built upon the inferred GRNs and RegFactors:
Regulatory Activity Inference: The activity of individual TFs or entire RegFactors is quantified from the collective expression of their target regulons (defined by \(G_{activation}\) or \(U^{(TG)}\)) using a multivariate linear model (MLM).
Intercellular Communication: scMagnify models signaling-to-transcription cascades by correlating the expression of receptors with the expression (or inferred activity) of their downstream TFs along the pseudotime axis. The significance of these interactions is validated using a permutation test to reveal how extracellular cues are translated into intracellular regulatory programs.
See “Decomposing multi-scale dynamic regulation from single-cell multiomics with scMagnify” for a detailed description of the methods and applications on different biological systems.