RegFactor Decomposition with scMagnify#
Preliminaries#
In this tutorial, you will learn how to:
RegFactor Decomposition. Decipher the combinatorial effects of transcription factors (TFs) using tensor decomposition.
Enrichment Analysis. Perform gene functional enrichment analysis based on the resulting Regulons.
The Rationale for Decomposition#
In the previous steps, we inferred a GRN and identified key individual regulators. However, it is well-established that TFs rarely act alone; they typically function in a combinatorial manner to orchestrate complex gene expression programs [].
To systematically explore these regulatory synergies, scMagnify implements a RegFactor Decomposition module. This approach is built upon a modified Tucker decomposition, a powerful technique from tensor analysis.
Instead of decomposing raw expression, this method is applied directly to a tensor constructed from the regulatory coefficients (Reg. scores) generated by our multi-scale Granger causality model. This allows the decomposition to focus purely on the inferred regulatory logic.
The primary goal is to simultaneously identify:
Co-regulating TF modules (groups of TFs that function as a unit).
Shared sets of Target Genes (TGs) that are controlled by these modules across different time lags.
This analysis elevates our understanding from individual TF-TG links to the higher-order “teams” of regulators driving the cellular process.
For a complete theoretical breakdown of the tensor decomposition method and the model mathematics, please refer to the [About the MSNGC](./About the MSNGC.md) section and the Methods section of our manuscript.
Import packages#
%load_ext autoreload
%autoreload 2
import warnings
from numba.core.errors import NumbaDeprecationWarning
warnings.simplefilter("ignore", category=NumbaDeprecationWarning)
warnings.simplefilter("ignore", FutureWarning)
warnings.simplefilter("ignore", UserWarning)
warnings.simplefilter("ignore", RuntimeWarning)
import os
import matplotlib.pyplot as plt
import scanpy as sc
import decoupler as dc
import scmagnify as scm
import scmagnify.logging as logg
from scmagnify.settings import settings
scm.info()
 |
|
Configurations#
scm.settings.verbosity = 2
%matplotlib inline
scm.settings.set_figure_params(
dpi=100,
facecolor="white",
frameon=False,
)
scm.load_fonts(["Arial"])
plt.rcParams["font.family"] = "Arial"
plt.rcParams["grid.alpha"] = 0
# Setting a workspace
dirPjtHome = "/mnt/TrueNas/project/chenxufeng/Data/PMID36973557_NatBiotechnol2023_T-cell-depleted/"
workDir = os.path.join(dirPjtHome, "scmagnify_wd")
scm.set_workspace(workDir)
workspace: /mnt/TrueNas/project/chenxufeng/Data/PMID36973557_NatBiotechnol2023_T-cell-depleted/scmagnify_wd/ ├── data ├── models ├── tmpfiles └── figures
# Set up Reference Genome
scm.set_genome(version="hg38", genomes_dir="/home/chenxufeng/picb_cxf/Ref/human/hg38/")
Genome Information ┏━━━━━━━━━┳━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓ ┃ Version ┃ Provider ┃ Directory ┃ ┡━━━━━━━━━╇━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩ │ hg38 │ UCSC │ /home/chenxufeng/picb_cxf/Ref/human/hg38/ │ └─────────┴──────────┴───────────────────────────────────────────┘
Load the data#
gdata = scm.read(os.path.join(settings.data_dir, "t-cell-depleted-bm_NaiveB_final.h5mu"))
RegFactor Decomposition#
The decomposition workflow is a straightforward, multi-step process.
First, we initialize the RegDecomp object from our gdata. This class automatically extracts the regulatory coefficient tensor and prepares it for decomposition.
decomper = scm.RegDecomp(gdata)
INFO Initializing RegDecomp object with multiscale mode.
INFO Filtering Network: Method: quantile Parameter: 0.0 Binarize: False
Next, we run the tucker_decomposition() method. This is the core step where the tensor is factorized. The rank parameter is critical, as it defines the number of components (RegFactors) to identify. The log output confirms the tensor shape and the Normalized Reconstruction Error (NRE), which measures how well the decomposition approximates the original data.
decomper.tucker_decomposition(rank=5)
INFO RegFactor Decomposing with Tucker decomposition: Tensor shape: (5, 105, 2926) Rank: 5
INFO Decomposition NRE: 0.82133766130817
Once the RegFactors (TF modules and their TG sets) are defined, we compute an activity score for each factor in each cell. This is done using the decoupler mlm method, parallel to the individual TF activity calculation.
decomper.compute_activity()
By default, factors are named numerically. We can use rename_regfactors() to re-order or rename them for better biological interpretation in downstream plots.
decomper.rename_regfactors(
{
"RegFactor_1": "RegFactor_3",
"RegFactor_2": "RegFactor_5",
"RegFactor_3": "RegFactor_4",
"RegFactor_4": "RegFactor_1",
"RegFactor_5": "RegFactor_2",
},
sort=True,
)
INFO Sorting RegFactors alphabetically.
INFO Renaming RegFactors: {'RegFactor_1': 'RegFactor_3', 'RegFactor_2': 'RegFactor_5', 'RegFactor_3': 'RegFactor_4', 'RegFactor_4': 'RegFactor_1', 'RegFactor_5': 'RegFactor_2'}
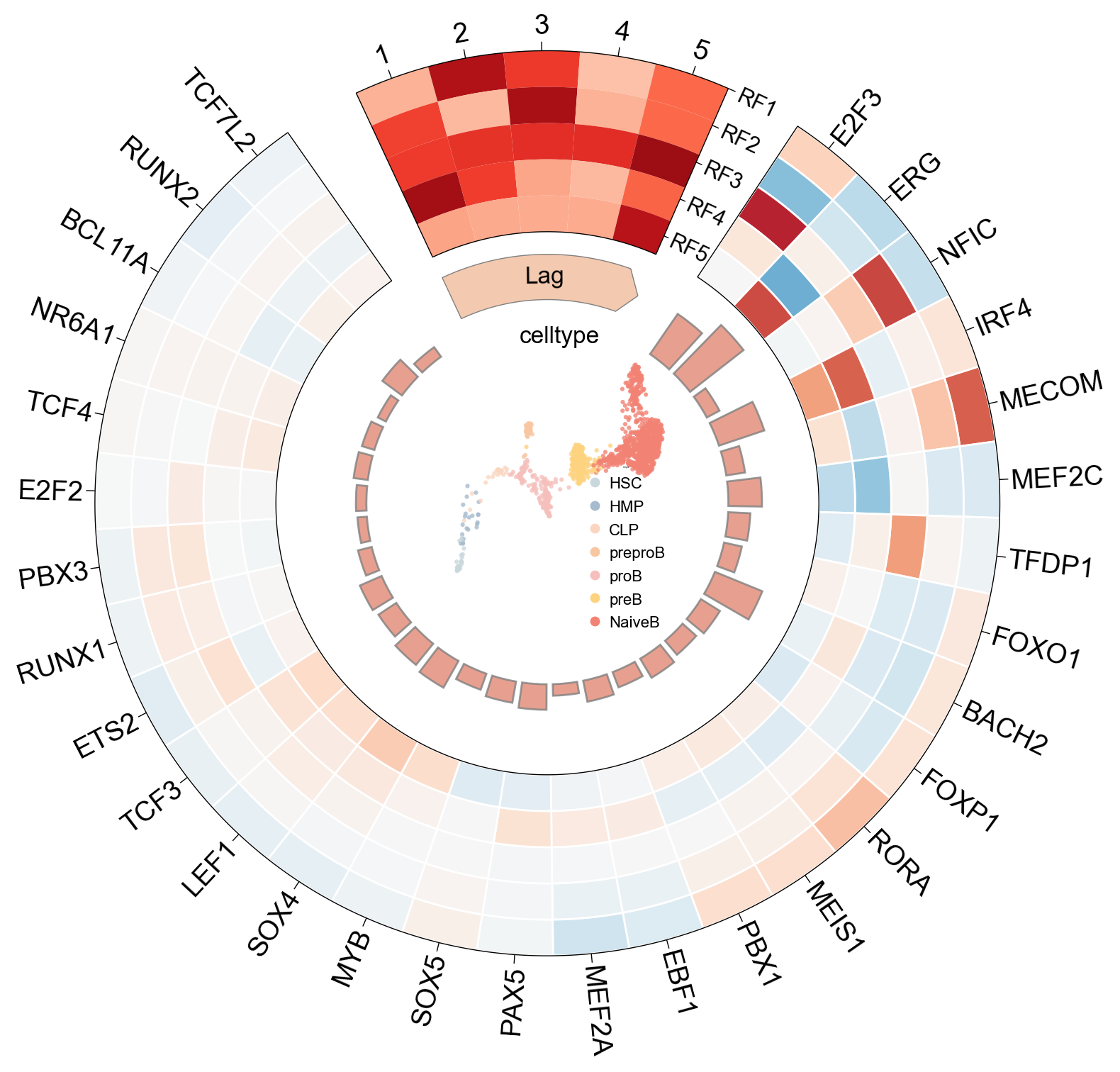
The circosplot() function provides a comprehensive, all-in-one visualization of the decomposition results. This plot effectively summarizes the key components of the inferred regulatory modules:
Outer Ring: The top transcription factors (TFs) that contribute most significantly to each RegFactor.
Inner Heatmap: The time lag associated with each RegFactor.
Center Plot: An embedding (specified by
embedding_key) colored by a cell annotation (specified bycolor_key).
scm.pl.circosplot(
gdata,
top_tfs=30,
cluster=True,
colorbar=False,
label_kws={"label_size": 14},
embedding_key="X_umap",
color_key="celltype",
center_axes_rect=(0.40, 0.46, 0.20, 0.20),
)
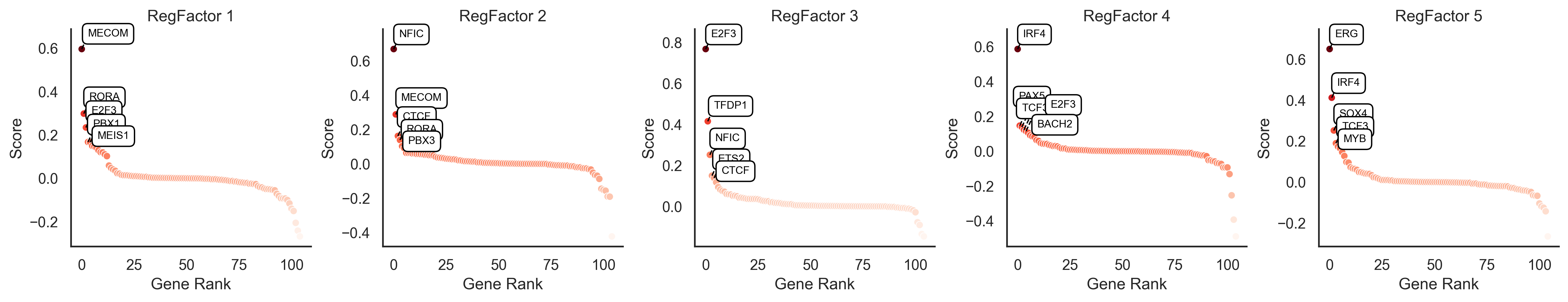
TF Loadings#
The “TF loadings” represent the contribution score of each transcription factor to a given RegFactor module. We can visualize these scores to understand the composition of each module.
scm.pl.rankplot(
gdata,
modal="RegFactor",
key="TF_loadings",
cmap="Reds",
n_top=5,
ncols=5,
wspace=0.3,
swap_df=True,
figsize=(20, 3),
show=True,
)
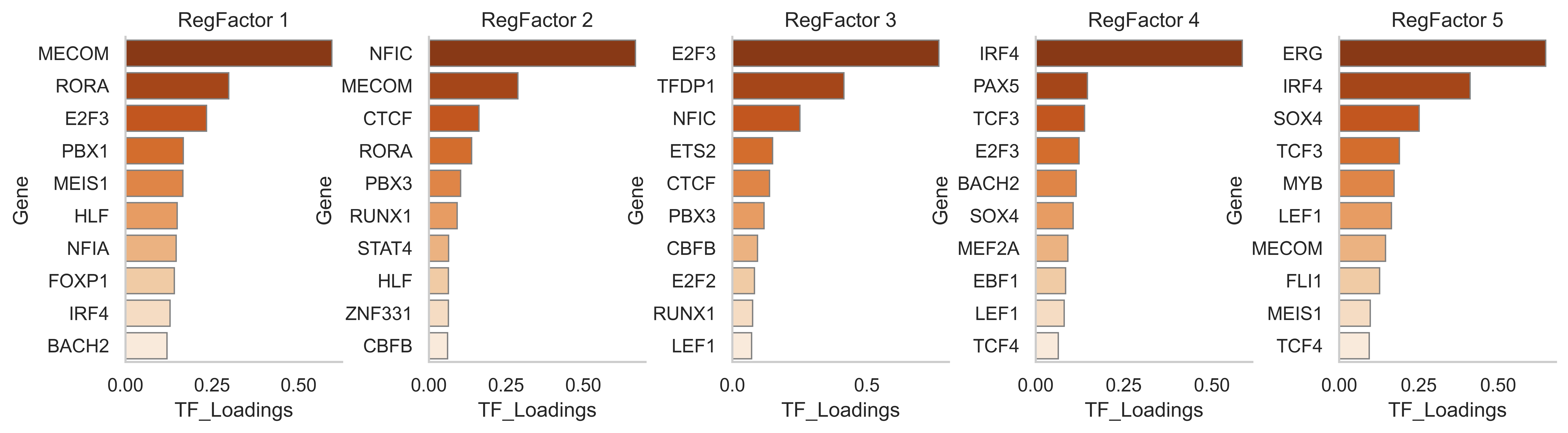
scm.pl.barplot(
gdata, modal="RegFactor", key="TF_loadings", swap_df=True, n_top=10, ncols=5, cmap="Oranges_r", xlabel="TF_Loadings"
)
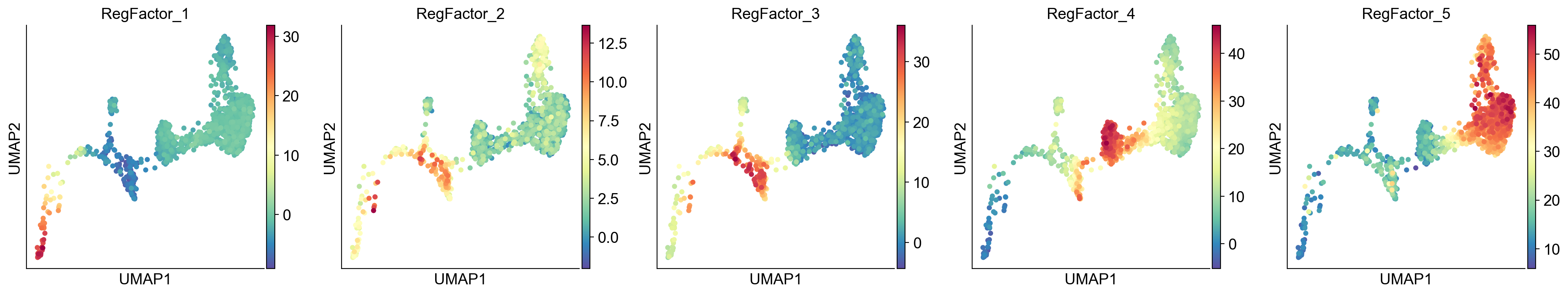
RegFactor Activity#
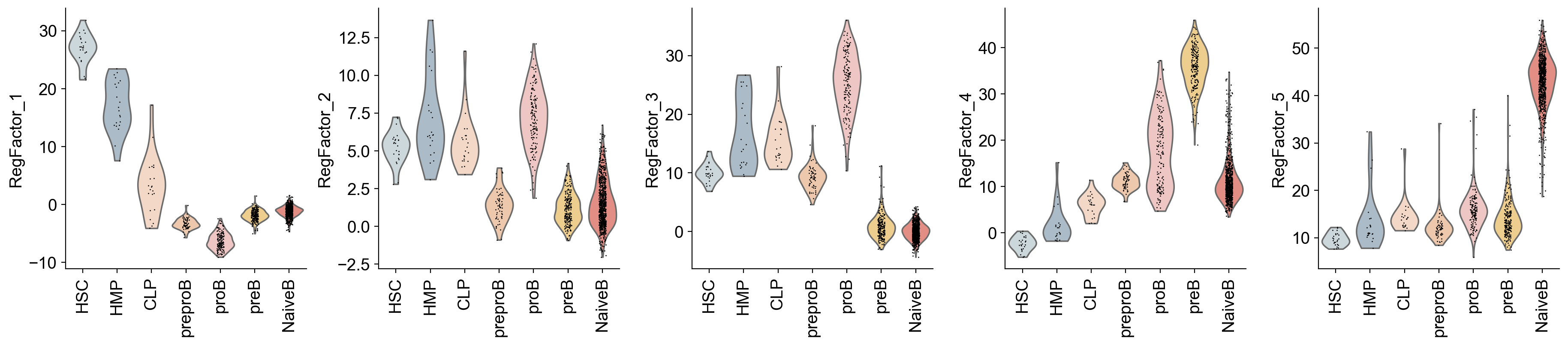
The activity of each RegFactor represents the combined regulatory influence of its constituent TFs. This activity is computed for each cell and stored in gdata["RegFactor"]. We can visualize these activities just like individual TF activities.
sc.pl.umap(gdata["RegFactor"], color=gdata["RegFactor"].var_names, ncols=5, cmap="Spectral_r")
sc.pl.violin(gdata["RegFactor"], groupby="celltype", keys=gdata["RegFactor"].var_names, rotation=90)
Finally, we can use scm.pl.trendplot to visualize the dynamic changes in RegFactor activity over pseudotime, allowing us to see when each regulatory module is turned “on” or “off” during the differentiation process.
scm.pl.trendplot(
gdata,
var_dict={
"RegFactor_1": [("RegFactor", None)],
"RegFactor_2": [("RegFactor", None)],
"RegFactor_3": [("RegFactor", None)],
"RegFactor_4": [("RegFactor", None)],
"RegFactor_5": [("RegFactor", None)],
},
normalize=True,
sortby="palantir_pseudotime",
col_color=["celltype"],
figsize=(6, 3),
dpi=100,
n_splines=5,
show_tkey=False,
show_stds=False,
)
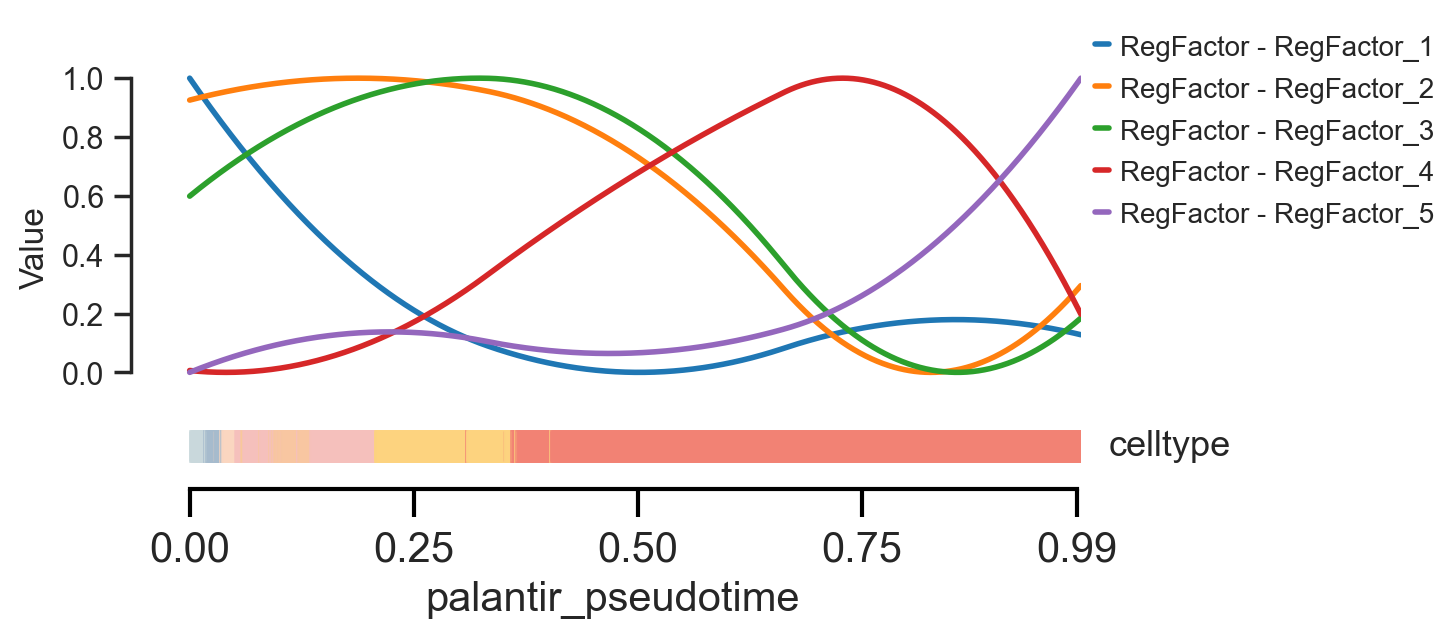
<Axes: xlabel='palantir_pseudotime', ylabel='Value'>
Funch Enrich#
To understand the biological functions of the identified RegFactors, we can perform a functional enrichment analysis on their target gene sets. scMagnify implements a standard Over-Representation Analysis (ORA) for this purpose.
First, we can define a color palette for our RegFactors, which will be used in subsequent plots.
gdata.uns["regfactors_colors"] = ["#CED7DA", "#AFBAC9", "#E6C8C3", "#E6CF8C", "#D49284"]
from scmagnify.tools import FuncEnrich
Next, we initialize the {py:class}~scmagnify.tools.FuncEnrich object. We specify the gene set database to use—in this case, the human MSigDB (v2025.1) gene symbols. We then filter this comprehensive database to include only the “Gene Ontology Biological Process” (GOBP) terms.
enricher = FuncEnrich(gene_sets="msigdb.v2025.1.Hs.symbols")
enricher.filter_genesets(pattern="GOBP")
INFO Initializing FuncEnrich object.
INFO Loading built-in gene set: msigdb.v2025.1.Hs.symbols from /home/chenxufeng/WorkSpace/Git-repos/scMagnify/src/scmagnify/data/genesets/msigdb.v2025.1.Hs.symbols.gmt
INFO Successfully loaded 35134 gene sets with a total of 43351 unique gene symbols.
INFO Filtering 35134 gene sets with pattern: 'GOBP'.
INFO Filter applied. Kept 7583 of 35134 gene sets.
tg_dict = scm.tl.extract_regfactor_genes(
gdata,
mode="TG",
plot=True,
n_top=500,
)
RegFactor Gene Summary ┏━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━┓ ┃ RegFactor ┃ Number of Genes ┃ ┡━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━┩ │ RegFactor_1 │ 500 │ │ RegFactor_2 │ 500 │ │ RegFactor_3 │ 500 │ │ RegFactor_4 │ 500 │ │ RegFactor_5 │ 500 │ └─────────────┴─────────────────┘
enr_pvals_dict = {}
regfactors_to_process = list(tg_dict.keys()) # Create a list to iterate over
for regfactor in regfactors_to_process:
logg.info(f"Running ORA for regfactor: [bold cyan]{regfactor}[/bold cyan]")
top_genes = tg_dict[regfactor].index
# Use the run_ora method from the initialized object.
# For best results, set n_background to the total number of genes
# detected in your experiment (e.g., len(adata.var_names)).
enr_pvals = enricher.run_ora(
gene_list=top_genes,
n_background=20000, # Recommended: Replace with your actual background size
top_n_results=10, # Show top 5 results in the console summary
)
# Store the full results
enr_pvals_dict[regfactor] = enr_pvals
# Check if the results DataFrame is not empty before plotting
if enr_pvals.empty:
logg.warning(f"No enrichment results found for {regfactor}. Skipping plot.")
continue
# Filter for significant results
enr_pvals_fil = enr_pvals[enr_pvals["FDR p-value"] < 0.05]
# Check if any significant terms remain after filtering
if enr_pvals_fil.empty:
logg.warning(f"No significant terms (FDR < 0.05) for {regfactor}. Skipping plot.")
continue
# Create the dot plot for the top 30 significant terms
dc.pl.dotplot(
enr_pvals_fil.sort_values("Combined score", ascending=False).head(30),
x="Combined score",
y="Term",
s="Odds ratio",
c="FDR p-value",
cmap="viridis",
scale=0.1,
figsize=(3, 6),
)
plt.title(regfactor)
plt.show()
INFO Running ORA for regfactor: RegFactor_1
INFO Starting ORA for a list of 500 genes.
INFO Using a user-defined background size of 20000 genes.
Top 10 Enriched Terms ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━┓ ┃ Term ┃ Overlap ┃ Combined score ┃ FDR p-value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━┩ │ GOBP_SPHINGOLIPID_TRANSLOCATION │ 4/5 │ 4.34e+02 │ 3.68e-04 │ │ GOBP_POSITIVE_REGULATION_OF_FC_RECEPTOR_MEDIATED_… │ 3/6 │ 1.76e+02 │ 1.31e-02 │ │ GOBP_EMBRYONIC_HEMOPOIESIS │ 7/24 │ 1.67e+02 │ 3.23e-04 │ │ GOBP_PLATELET_ACTIVATING_FACTOR_METABOLIC_PROCESS │ 3/8 │ 1.18e+02 │ 2.59e-02 │ │ GOBP_NEGATIVE_REGULATION_OF_SMOOTH_MUSCLE_CONTRAC… │ 4/14 │ 1.01e+02 │ 1.38e-02 │ │ GOBP_PRIMITIVE_HEMOPOIESIS │ 3/9 │ 1.00e+02 │ 3.18e-02 │ │ GOBP_LYMPHOCYTE_DIFFERENTIATION │ 39/440 │ 9.70e+01 │ 3.22e-08 │ │ GOBP_POSITIVE_REGULATION_OF_MAST_CELL_PROLIFERATI… │ 2/5 │ 9.35e+01 │ 9.62e-02 │ │ GOBP_NUCLEAR_MIGRATION_ALONG_MICROTUBULE │ 2/5 │ 9.35e+01 │ 9.62e-02 │ │ GOBP_REGULATION_OF_CELL_DIFFERENTIATION_INVOLVED_… │ 2/5 │ 9.35e+01 │ 9.62e-02 │ └────────────────────────────────────────────────────┴─────────┴────────────────┴─────────────┘

INFO Running ORA for regfactor: RegFactor_2
INFO Starting ORA for a list of 500 genes.
INFO Using a user-defined background size of 20000 genes.
Top 10 Enriched Terms ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━┓ ┃ Term ┃ Overlap ┃ Combined score ┃ FDR p-value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━┩ │ GOBP_POSITIVE_REGULATION_OF_FC_RECEPTOR_MEDIATED_… │ 4/6 │ 3.37e+02 │ 6.16e-04 │ │ GOBP_REGULATION_OF_FC_RECEPTOR_MEDIATED_STIMULATO… │ 4/11 │ 1.43e+02 │ 5.43e-03 │ │ GOBP_ESTABLISHMENT_OF_MITOTIC_SISTER_CHROMATID_CO… │ 3/7 │ 1.42e+02 │ 1.52e-02 │ │ GOBP_KINETOCHORE_ORGANIZATION │ 6/21 │ 1.41e+02 │ 7.85e-04 │ │ GOBP_KINETOCHORE_ASSEMBLY │ 5/17 │ 1.27e+02 │ 2.72e-03 │ │ GOBP_MAINTENANCE_OF_LENS_TRANSPARENCY │ 3/8 │ 1.18e+02 │ 2.12e-02 │ │ GOBP_NUCLEAR_BODY_ORGANIZATION │ 5/18 │ 1.16e+02 │ 3.39e-03 │ │ GOBP_THYMIC_T_CELL_SELECTION │ 6/24 │ 1.15e+02 │ 1.63e-03 │ │ GOBP_NEGATIVE_T_CELL_SELECTION │ 4/13 │ 1.12e+02 │ 9.04e-03 │ │ GOBP_MITOTIC_SISTER_CHROMATID_COHESION │ 7/31 │ 1.12e+02 │ 7.84e-04 │ └────────────────────────────────────────────────────┴─────────┴────────────────┴─────────────┘

INFO Running ORA for regfactor: RegFactor_3
INFO Starting ORA for a list of 500 genes.
INFO Using a user-defined background size of 20000 genes.
Top 10 Enriched Terms ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━┓ ┃ Term ┃ Overlap ┃ Combined score ┃ FDR p-value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━┩ │ GOBP_CRD_MEDIATED_MRNA_STABILIZATION │ 5/11 │ 2.40e+02 │ 2.54e-04 │ │ GOBP_CELL_CYCLE_PROCESS │ 105/1348 │ 2.17e+02 │ 1.29e-22 │ │ GOBP_POSITIVE_REGULATION_OF_CYTOPLASMIC_TRANSLATI… │ 6/17 │ 1.95e+02 │ 1.65e-04 │ │ GOBP_MITOTIC_CELL_CYCLE_PROCESS │ 70/783 │ 1.85e+02 │ 1.47e-17 │ │ GOBP_POSITIVE_REGULATION_OF_PROTEIN_LOCALIZATION_… │ 3/6 │ 1.76e+02 │ 8.46e-03 │ │ GOBP_POSITIVE_REGULATION_OF_MAINTENANCE_OF_SISTER… │ 3/6 │ 1.76e+02 │ 8.46e-03 │ │ GOBP_CELL_CYCLE │ 116/1705 │ 1.74e+02 │ 1.08e-20 │ │ GOBP_MITOTIC_CELL_CYCLE │ 75/931 │ 1.57e+02 │ 1.75e-16 │ │ GOBP_POSITIVE_REGULATION_OF_CELL_CYCLE_PROCESS │ 33/275 │ 1.54e+02 │ 2.72e-11 │ │ GOBP_POSITIVE_REGULATION_OF_CELL_CYCLE │ 38/346 │ 1.49e+02 │ 8.71e-12 │ └────────────────────────────────────────────────────┴──────────┴────────────────┴─────────────┘

INFO Running ORA for regfactor: RegFactor_4
INFO Starting ORA for a list of 500 genes.
INFO Using a user-defined background size of 20000 genes.
Top 10 Enriched Terms ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━┓ ┃ Term ┃ Overlap ┃ Combined score ┃ FDR p-value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━┩ │ GOBP_DNA_TOPOLOGICAL_CHANGE │ 5/10 │ 2.76e+02 │ 1.54e-04 │ │ GOBP_DNA_CONFORMATION_CHANGE │ 6/18 │ 1.79e+02 │ 2.27e-04 │ │ GOBP_POSITIVE_REGULATION_OF_FC_RECEPTOR_MEDIATED_… │ 3/6 │ 1.76e+02 │ 7.69e-03 │ │ GOBP_POSITIVE_REGULATION_OF_GAMMA_DELTA_T_CELL_AC… │ 4/11 │ 1.43e+02 │ 3.62e-03 │ │ GOBP_B_CELL_RECEPTOR_SIGNALING_PATHWAY │ 14/78 │ 1.42e+02 │ 1.64e-06 │ │ GOBP_REGULATION_OF_NUCLEOTIDE_EXCISION_REPAIR │ 7/29 │ 1.24e+02 │ 3.39e-04 │ │ GOBP_REGULATION_OF_CELLULAR_PH_REDUCTION │ 3/8 │ 1.18e+02 │ 1.61e-02 │ │ GOBP_POSITIVE_REGULATION_OF_CHROMOSOME_CONDENSATI… │ 3/8 │ 1.18e+02 │ 1.61e-02 │ │ GOBP_DNA_GEOMETRIC_CHANGE │ 3/8 │ 1.18e+02 │ 1.61e-02 │ │ GOBP_B_CELL_DIFFERENTIATION │ 20/149 │ 1.17e+02 │ 2.82e-07 │ └────────────────────────────────────────────────────┴─────────┴────────────────┴─────────────┘

INFO Running ORA for regfactor: RegFactor_5
INFO Starting ORA for a list of 500 genes.
INFO Using a user-defined background size of 20000 genes.
Top 10 Enriched Terms ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━┳━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━┓ ┃ Term ┃ Overlap ┃ Combined score ┃ FDR p-value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━╇━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━┩ │ GOBP_B_CELL_RECEPTOR_SIGNALING_PATHWAY │ 23/78 │ 5.20e+02 │ 8.95e-16 │ │ GOBP_PEPTIDE_ANTIGEN_ASSEMBLY_WITH_MHC_CLASS_II_P… │ 8/16 │ 4.24e+02 │ 1.64e-07 │ │ GOBP_PEPTIDE_ANTIGEN_ASSEMBLY_WITH_MHC_PROTEIN_CO… │ 8/21 │ 2.83e+02 │ 1.64e-06 │ │ GOBP_ANTIGEN_RECEPTOR_MEDIATED_SIGNALING_PATHWAY │ 34/214 │ 2.70e+02 │ 3.64e-15 │ │ GOBP_REGULATION_OF_B_CELL_PROLIFERATION │ 16/68 │ 2.53e+02 │ 1.06e-09 │ │ GOBP_LYMPHOCYTE_ACTIVATION │ 80/846 │ 2.44e+02 │ 1.47e-21 │ │ GOBP_POSITIVE_REGULATION_OF_B_CELL_PROLIFERATION │ 12/47 │ 2.21e+02 │ 1.33e-07 │ │ GOBP_ANTIGEN_PROCESSING_AND_PRESENTATION_OF_EXOGE… │ 11/41 │ 2.20e+02 │ 3.28e-07 │ │ GOBP_REGULATION_OF_B_CELL_RECEPTOR_SIGNALING_PATH… │ 8/25 │ 2.17e+02 │ 6.51e-06 │ │ GOBP_ANTIGEN_PROCESSING_AND_PRESENTATION_OF_EXOGE… │ 9/31 │ 2.08e+02 │ 3.05e-06 │ └────────────────────────────────────────────────────┴─────────┴────────────────┴─────────────┘

Network Visualization#
from scmagnify.plotting import GRNVisualizer
custom_term_dict = {
"GOBP_B_CELL_RECEPTOR_SIGNALING_PATHWAY": [
"LYN",
"PTPRC",
"IGHM",
"PRKCB",
"BLK",
"IGKC",
"GRB2",
"FOXP1",
"NFKB1",
],
"GOBP_B_CELL_DIFFERENTIATION": ["PAX5", "PTPRJ", "PTPRC", "IKZF3", "IGHM", "HDAC9", "CARD11"],
"GOBP_CHROMATIN_REMODELING": [
"CHD7",
"NCOA3",
"HDAC9",
"SMCHD1",
"PRKCB",
"ATAD2",
"SETBP1",
"PCGF5",
"LBR",
"DPF3",
],
"GOBP_DNA_TOPOLOGICAL_CHANGE": ["TOP2A", "TOP2B", "HGMB1", "HGMB2"],
}
tf_list = ["EBF1", "PAX5", "IRF4", "TCF3", "SOX4"]
tg_list = tg_list = (
gdata["RegFactor"].varm["TG_loadings"].T.sort_values(by="RegFactor_4", ascending=False).head(100).index
)
# get tg from custom terms
tg_list = []
for _term, genes in custom_term_dict.items():
tg_list.extend(genes)
vis = GRNVisualizer(gdata)
vis.prepare_network(regulon=None, tf_list=tf_list, tg_list=tg_list, add_suffixes=True, target_clusters=custom_term_dict)
INFO Building base TF-peak-gene triplet network...
INFO Filtering triplet network using 'network' from gdata.uns...
INFO Base network constructed with 91074 TF-peak-gene relationships.
INFO Filtering network based on provided TF and/or Target Gene lists...
INFO Final network contains 155 relationships after filtering.
INFO Using provided manual target gene clusters.
Manual Target Gene Clusters Summary ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━┓ ┃ Cluster ┃ Num Genes ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━┩ │ GOBP_B_CELL_RECEPTOR_SIGNALING_PATHWAY │ 8 │ │ GOBP_B_CELL_DIFFERENTIATION │ 6 │ │ GOBP_CHROMATIN_REMODELING │ 10 │ └────────────────────────────────────────┴───────────┘
<scmagnify.plotting._grn_visualizer.GRNVisualizer at 0x7f44800e48b0>
vis.add_continuous_mapping(
node_type="TF",
visual_property="size",
modal="GRN", # Assumes gdata has a modal named 'activity'
varm_key=("network_score", "degree_centrality"),
map_range=(8, 12), # Map sizes from 8 to 25
legend_title="Degree Centrality",
)
INFO Added continuous mapping for 'TF' node 'size'.
<scmagnify.plotting._grn_visualizer.GRNVisualizer at 0x7f44800e48b0>
vis.add_continuous_mapping(
node_type="Target",
visual_property="color",
modal="RNA", # Assumes gdata has a modal named 'activity'
map_range="Reds",
layer="log1p_norm",
legend_title="RNA Expression",
)
INFO Added continuous mapping for 'Target' node 'color'.
<scmagnify.plotting._grn_visualizer.GRNVisualizer at 0x7f44800e48b0>
vis.r_tf = 0.5
vis.r_ccre = 0.8
vis.r_target = 1.3
vis.plot(
figsize=(5, 5),
dpi=300,
title="RegFactor 4 eGRN",
title_fontsize=12,
node_label_offset=0.2,
node_edge_width=1,
tf_layout_mode="uniform",
label_offset_factor=1,
label_mode="external",
edge_layout="bundled",
highlight={"Target": ["PAX5"], "cCRE": ["chr9:37029184-37029684"], "TF": ["EBF1", "TCF3", "PAX5", "IRF4", "SOX4"]},
interactive=False,
jitter_strength=5,
draw_cluster_wedges=False,
)
INFO Calculating Network Layout...
INFO Applying highlighting to specified nodes and edges...
INFO Applying custom continuous mappings...
Adjusting 21 external target labels to prevent overlap...
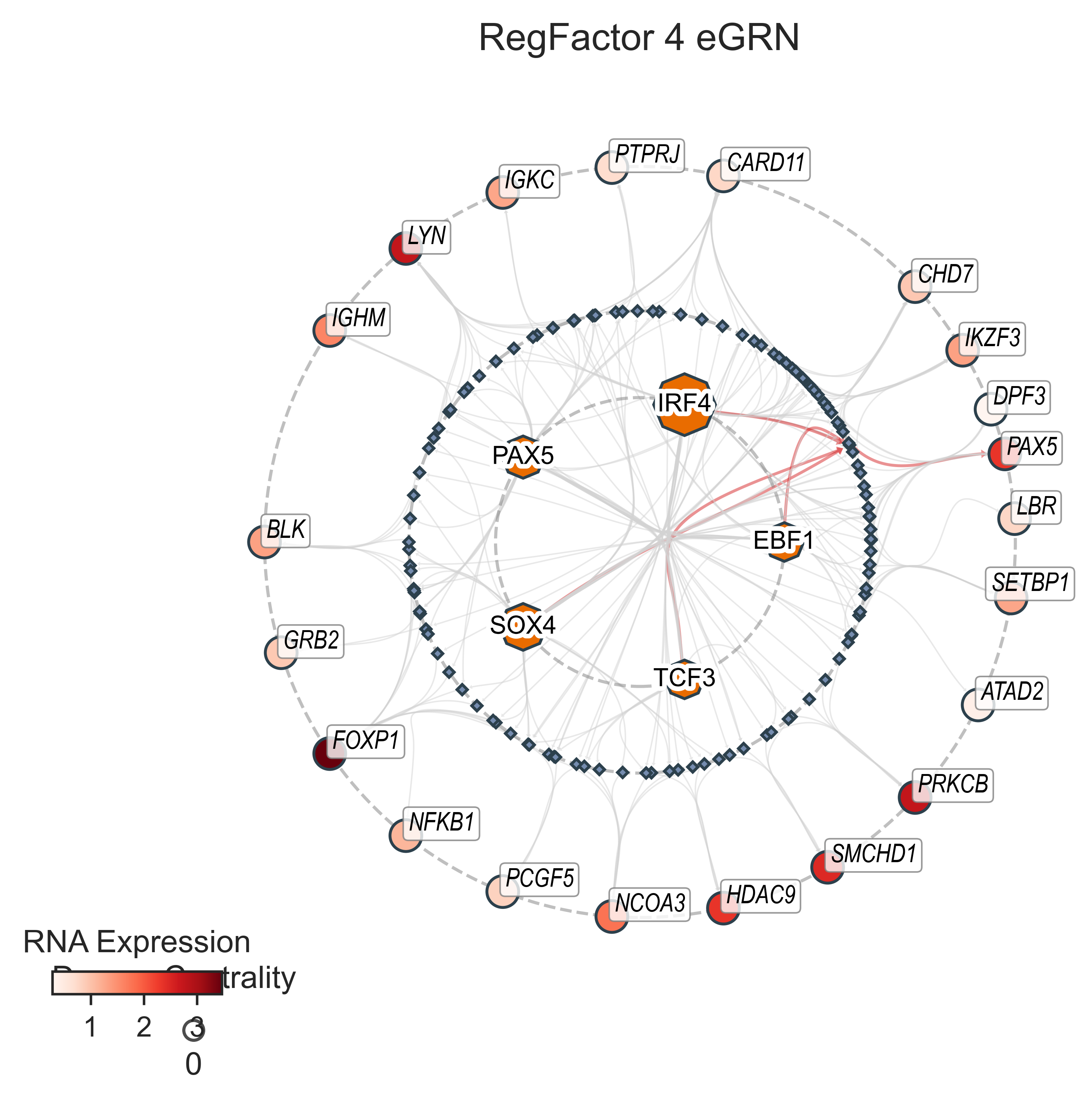
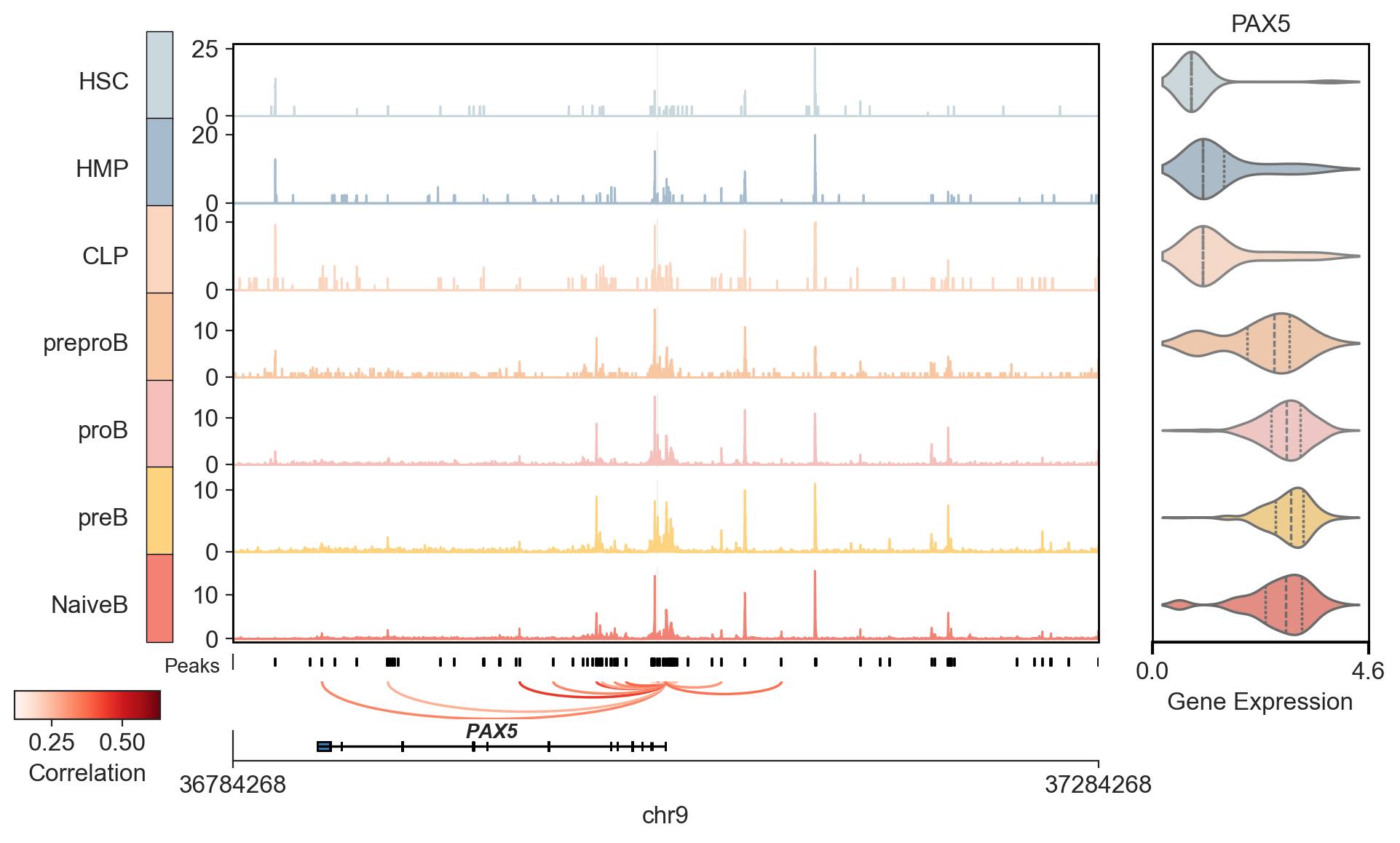
Genomic Viewer#
from scmagnify.plotting import GenomeViewer
fragment_files = {
"IM-1393_BoneMarrow_TcellDep_1_multiome": "/mnt/TrueNas/project/chenxufeng//Data/PMID36973557_NatBiotechnol2023_T-cell-depleted/0_cellranger/BM_Tcelldep_Rep1_atac_fragments.tsv.gz",
"IM-1393_BoneMarrow_TcellDep_2_multiome": "/mnt/TrueNas/project/chenxufeng//Data/PMID36973557_NatBiotechnol2023_T-cell-depleted/0_cellranger/BM_Tcelldep_Rep2_atac_fragments.tsv.gz",
}
viewer = GenomeViewer(data=gdata, modal="ATAC", fragment_files=fragment_files, cluster="celltype")
INFO Attempting to auto-load peaks from 'ATAC' modality's .var_names...
INFO ✅ Successfully auto-loaded 216477 peaks as 'Peaks'.
INFO Attempting to auto-load links from gdata.uns['peak_gene_corrs']['filtered_corrs']...
INFO ✅ Successfully auto-loaded 22745 links from gdata.
INFO Preprocessing metadata...
INFO Loading and processing GTF...
viewer.plot(
anchor_gene="PAX5",
theme="ticks",
side_genes=["PAX5"],
anchor_flank=250000,
normalize=True,
show_motif_logos=False,
rasterize_coverage=True,
highlight_peaks=["chr9:37029184-37029684"],
)
INFO Processing peaks for highlighting...
INFO peak gene cor pval chrom start \ 5731 chr9:36835204-36835704 PAX5 0.417219 0.002627 chr9 36835204 5732 chr9:36873230-36873730 PAX5 0.280841 0.045795 chr9 36873230 5733 chr9:36949396-36949896 PAX5 0.633355 0.001788 chr9 36949396 5734 chr9:36968883-36969383 PAX5 0.407278 0.007072 chr9 36968883 5735 chr9:36993858-36994358 PAX5 0.586656 0.009581 chr9 36993858 5736 chr9:36997154-36997654 PAX5 0.304038 0.022332 chr9 36997154 5737 chr9:37004392-37004892 PAX5 0.397404 0.007143 chr9 37004392 5738 chr9:37010921-37011421 PAX5 0.513678 0.002850 chr9 37010921 5739 chr9:37025579-37026079 PAX5 0.132025 0.086116 chr9 37025579 5740 chr9:37029184-37029684 PAX5 0.443045 0.018797 chr9 37029184 5741 chr9:37030276-37030776 PAX5 0.118639 0.063281 chr9 37030276 5742 chr9:37032656-37033156 PAX5 0.383195 0.002373 chr9 37032656 5743 chr9:37034085-37034585 PAX5 0.404153 0.029794 chr9 37034085 5744 chr9:37037198-37037698 PAX5 0.137647 0.090633 chr9 37037198 5745 chr9:37037840-37038340 PAX5 0.403106 0.007047 chr9 37037840 5746 chr9:37040435-37040935 PAX5 0.248222 0.039122 chr9 37040435 5747 chr9:37066033-37066533 PAX5 0.362238 0.014985 chr9 37066033 5748 chr9:37100720-37101220 PAX5 0.475527 0.001061 chr9 37100720 end 5731 36835704 5732 36873730 5733 36949896 5734 36969383 5735 36994358 5736 36997654 5737 37004892 5738 37011421 5739 37026079 5740 37029684 5741 37030776 5742 37033156 5743 37034585 5744 37037698 5745 37038340 5746 37040935 5747 37066533 5748 37101220
INFO Plotting 18 links.
INFO Drawing highlights for 1 regions.
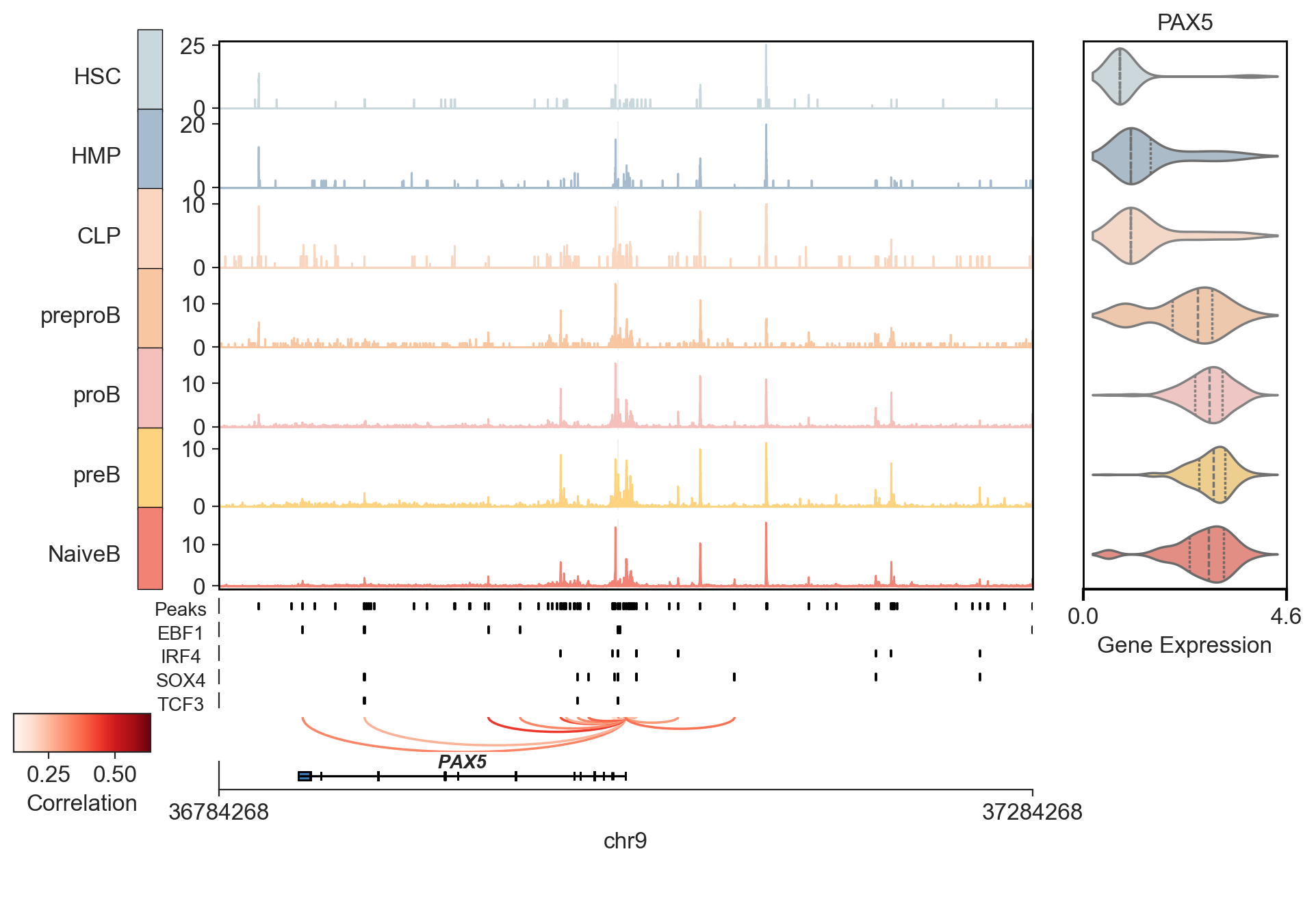
viewer.add_motifs(
tfs=["EBF1", "IRF4", "SOX4", "TCF3"],
motif_db="HOCOMOCOv11_HUMAN",
)
INFO Added 4 new motifs. Total motifs: 4.
INFO Added 4 motifs for plotting.
viewer.plot(
anchor_gene="PAX5",
theme="ticks",
side_genes=["PAX5"],
anchor_flank=250000,
normalize=True,
highlight_peaks=["chr9:37029184-37029684"],
show_motif_logos=False,
rasterize_coverage=True,
)
INFO Processing peaks for highlighting...
INFO peak gene cor pval chrom start \ 5731 chr9:36835204-36835704 PAX5 0.417219 0.002627 chr9 36835204 5732 chr9:36873230-36873730 PAX5 0.280841 0.045795 chr9 36873230 5733 chr9:36949396-36949896 PAX5 0.633355 0.001788 chr9 36949396 5734 chr9:36968883-36969383 PAX5 0.407278 0.007072 chr9 36968883 5735 chr9:36993858-36994358 PAX5 0.586656 0.009581 chr9 36993858 5736 chr9:36997154-36997654 PAX5 0.304038 0.022332 chr9 36997154 5737 chr9:37004392-37004892 PAX5 0.397404 0.007143 chr9 37004392 5738 chr9:37010921-37011421 PAX5 0.513678 0.002850 chr9 37010921 5739 chr9:37025579-37026079 PAX5 0.132025 0.086116 chr9 37025579 5740 chr9:37029184-37029684 PAX5 0.443045 0.018797 chr9 37029184 5741 chr9:37030276-37030776 PAX5 0.118639 0.063281 chr9 37030276 5742 chr9:37032656-37033156 PAX5 0.383195 0.002373 chr9 37032656 5743 chr9:37034085-37034585 PAX5 0.404153 0.029794 chr9 37034085 5744 chr9:37037198-37037698 PAX5 0.137647 0.090633 chr9 37037198 5745 chr9:37037840-37038340 PAX5 0.403106 0.007047 chr9 37037840 5746 chr9:37040435-37040935 PAX5 0.248222 0.039122 chr9 37040435 5747 chr9:37066033-37066533 PAX5 0.362238 0.014985 chr9 37066033 5748 chr9:37100720-37101220 PAX5 0.475527 0.001061 chr9 37100720 end 5731 36835704 5732 36873730 5733 36949896 5734 36969383 5735 36994358 5736 36997654 5737 37004892 5738 37011421 5739 37026079 5740 37029684 5741 37030776 5742 37033156 5743 37034585 5744 37037698 5745 37038340 5746 37040935 5747 37066533 5748 37101220
INFO Plotting 18 links.
INFO Drawing highlights for 1 regions.
Save the Data#
gdata.write(os.path.join(workDir, "gdata_tcelldep-bm_04_NaiveB.h5mu"))
Closing matters#
What’s next?#
In this tutorial, you learned how to use RegDecomp to perform tensor decomposition, identifying combinatorial RegFactors (TF modules) and their target genes. You also learned how to visualize these modules, analyze their activity, and interpret their biological function using enrichment analysis.
This completes the core scMagnify workflow. For your next steps, we recommend:
Applying this workflow to your own data. You can now use this four-part framework to analyze your own single-cell multi-omic datasets from end to end.
Refer to the API to explore all available functions and advanced parameter values that can be used to customize these computations for your specific biological questions.
If you encounter any bugs or have suggestions for new features, please open an issue. For general questions, please post on the scverse discourse or contact us at chenxufeng2022@sinh.ac.cn.
Package versions#
import session_info
session_info.show()
Click to view session information
----- matplotlib 3.8.4 numba 0.59.1 numpy 1.26.4 pandas 1.5.3 scanpy 1.10.1 scmagnify NA seaborn 0.13.2 session_info 1.0.0 -----
Click to view modules imported as dependencies
Bio 1.83 MOODS NA PIL 10.3.0 PyComplexHeatmap 1.8.1 absl NA adjustText 0.8 anndata 0.9.2 anyio NA appdirs 1.4.4 argcomplete NA arrow 1.3.0 asttokens NA attr 23.2.0 attrs 23.2.0 babel 2.15.0 biothings_client 0.3.1 brotli 1.1.0 cellrank 2.0.4 certifi 2024.08.30 cffi 1.16.0 chardet 5.2.0 charset_normalizer 3.3.2 chex 0.1.82 cloudpickle 3.0.0 colorama 0.4.6 comm 0.2.2 contextlib2 NA cycler 0.12.1 cython_runtime NA dateutil 2.9.0.post0 debugpy 1.8.1 decorator 5.1.1 defusedxml 0.7.1 deprecated 1.2.14 dill 0.3.8 diskcache 5.6.3 docrep 0.3.2 etils 1.5.2 exceptiongroup 1.2.1 executing 2.0.1 fastjsonschema NA filelock 3.14.0 flax 0.7.2 fontTools 4.51.0 fqdn NA fsspec 2024.5.0 genomepy 0.16.1 gmpy2 2.1.5 h5py 3.9.0 idna 3.7 igraph 0.11.8 importlib_metadata NA importlib_resources NA ipykernel 6.29.4 ipywidgets 8.1.5 isoduration NA jax 0.4.13 jaxlib 0.4.13 jedi 0.19.1 jinja2 3.0.3 joblib 1.4.2 json5 0.10.0 jsonpointer 3.0.0 jsonschema 4.22.0 jsonschema_specifications NA jupyter_events 0.10.0 jupyter_server 2.14.2 jupyterlab_server 2.27.3 kiwisolver 1.4.5 legacy_api_wrap NA leidenalg 0.10.2 lightning 2.1.4 lightning_utilities 0.11.2 llvmlite 0.42.0 loguru 0.7.2 louvain 0.8.2 lxml 5.3.0 markupsafe 2.1.5 matplotlib_inline 0.1.7 ml_collections NA ml_dtypes 0.4.0 mpl_toolkits NA mpmath 1.3.0 msgpack 1.0.8 mudata 0.2.3 multipledispatch 0.6.0 mygene 3.2.2 mysql NA natsort 8.4.0 nbformat 5.10.4 ncls 0.0.68 networkx 3.1 norns NA numexpr 2.10.0 numpyro 0.12.1 nvfuser NA opt_einsum v3.3.0 optax 0.2.1 overrides NA packaging 24.0 palettable 3.3.3 parso 0.8.4 patsy 0.5.6 petsc4py 3.19.1 pexpect 4.9.0 pkg_resources NA platformdirs 4.2.2 plotly 5.24.1 progressbar 4.4.2 prometheus_client NA prompt_toolkit 3.0.36 psutil 5.9.8 ptyprocess 0.7.0 pure_eval 0.2.2 pyarrow 18.0.0 pycirclize 1.7.2 pycparser 2.22 pydev_ipython NA pydevconsole NA pydevd 2.9.5 pydevd_file_utils NA pydevd_plugins NA pydevd_tracing NA pyfaidx 0.7.2.2 pygam 0.9.1 pygments 2.18.0 pygpcca 1.0.4 pyparsing 3.1.2 pyranges 0.1.1 pyro 1.9.0+f02dfb9 pysam 0.22.1 python_utils NA pythonjsonlogger NA pytz 2024.2 referencing NA requests 2.31.0 rfc3339_validator 0.1.4 rfc3986_validator 0.1.1 rich NA rpds NA scipy 1.12.0 scvelo 0.3.3.dev3+gf21651c scvi 1.1.6 send2trash NA six 1.16.0 sklearn 1.5.2 slepc4py 3.19.1 sniffio 1.3.1 socks 1.7.1 sorted_nearest 0.0.39 sparse 0.15.4 sphinxcontrib NA stack_data 0.6.3 statsmodels 0.14.2 swig_runtime_data4 NA sympy 1.12 tensorly 0.9.0 texttable 1.7.0 threadpoolctl 3.5.0 toolz 0.12.1 torch 2.0.0+cu118 torchmetrics 1.4.0.post0 tornado 6.4 tqdm 4.66.4 traitlets 5.14.3 typing_extensions NA uri_template NA urllib3 2.2.1 wcwidth 0.2.13 webcolors NA websocket 1.8.0 wrapt 1.16.0 yaml 6.0.1 zipp NA zmq 26.0.3 zoneinfo NA
----- IPython 8.18.0 jupyter_client 8.6.1 jupyter_core 5.7.2 jupyterlab 4.3.3 notebook 7.3.1 ----- Python 3.9.19 | packaged by conda-forge | (main, Mar 20 2024, 12:50:21) [GCC 12.3.0] Linux-3.10.0-1160.90.1.el7.x86_64-x86_64-with-glibc2.17 ----- Session information updated at 2025-02-17 00:54